
 Solving a mystery that stumped scientists for decades, last November a group of computational biologists from Alphabet’s DeepMind used AI to predict a protein’s structure from its amino acid sequence. Not even a year later, a new study offers a more powerful model, capable of computing protein structures in as little as 10 minutes, on … Continued
Solving a mystery that stumped scientists for decades, last November a group of computational biologists from Alphabet’s DeepMind used AI to predict a protein’s structure from its amino acid sequence. Not even a year later, a new study offers a more powerful model, capable of computing protein structures in as little as 10 minutes, on … Continued
Solving a mystery that stumped scientists for decades, last November a group of computational biologists from Alphabet’s DeepMind used AI to predict a protein’s structure from its amino acid sequence.
Not even a year later, a new study offers a more powerful model, capable of computing protein structures in as little as 10 minutes, on one gaming computer.
The research, from scientists at the University of Washington (UW), holds promise for faster drug development, which could unlock solutions for treating diseases like cancer.
Present in every cell in the body, proteins play a role in many processes such as blood clotting, hormone regulation, immune system response, vision, and cell and tissue repair. Made from long chains of amino acids that interact to form a folded three-dimensional structure, the shape of a protein determines its function.
Unfolded or misfolded proteins are also thought to cause degenerative disorders including cystic fibrosis, Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease. Understanding and predicting how a protein structure develops could help scientists design effective interventions for many of these diseases.
The researchers at UW developed the RoseTTAFold model by creating a three-track neural network that simultaneously considers the sequence patterns, amino acid interaction, and possible three-dimensional structure of a protein.
To train the model, the team used discontinuous crops of protein segments, with 260 unique amino acid elements. With the cuDNN-accelerated PyTorch deep learning framework, and NVIDIA GeForce 2080 GPUs, this information flows back and forth within the deep learning model. The network is then able to deduce a protein’s chemical parts along with its folded structure.
“The end-to-end version of RoseTTAFold requires about 10 minutes on an RTX 2080 GPU to generate backbone coordinates for proteins with less than 400 residues. The pyRosetta version requires 5 minutes for network calculations on a single NVIDIA RTX 2080 GPU, and an hour for all-atom structure generation with 15 CPU cores,” the researchers write in the study.
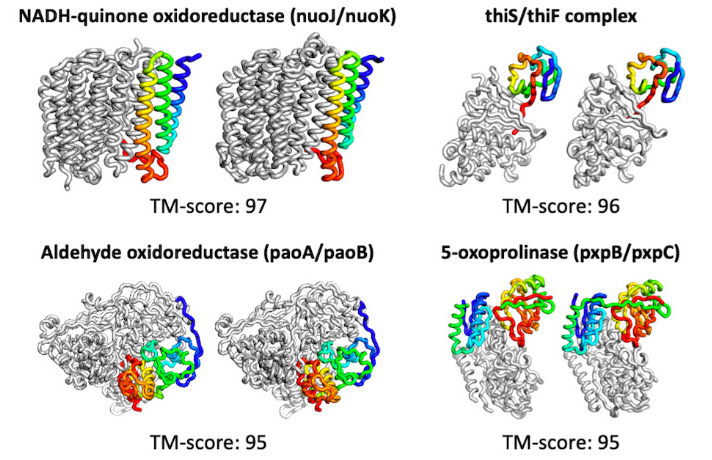
The tool not only quickly predicts proteins, but can do so with limited input. It also has the ability to compute beyond simple structures, predicting complexes consisting of several proteins bound together. More complex models are computed in about 30 minutes on a 24G NVIDIA TITAN RTX.
A public server is available for anyone interested in submitting protein sequences. The source code is also freely available to the scientific community.
“In just the last month, over 4,500 proteins have been submitted to our new web server, and we have made the RoseTTAFold code available through the GitHub website. We hope this new tool will continue to benefit the entire research community,” said lead author Minkyung Baek, a postdoctoral scholar at the University of Washington, Institute for Protein Design.