
 The GPU-accelerated Clara Parabricks v3.7 release brings support for gene panels, RNA-Seq, short tandem repeats, and updates to GATK 4.2 and DeepVariant 1.1.
The GPU-accelerated Clara Parabricks v3.7 release brings support for gene panels, RNA-Seq, short tandem repeats, and updates to GATK 4.2 and DeepVariant 1.1.
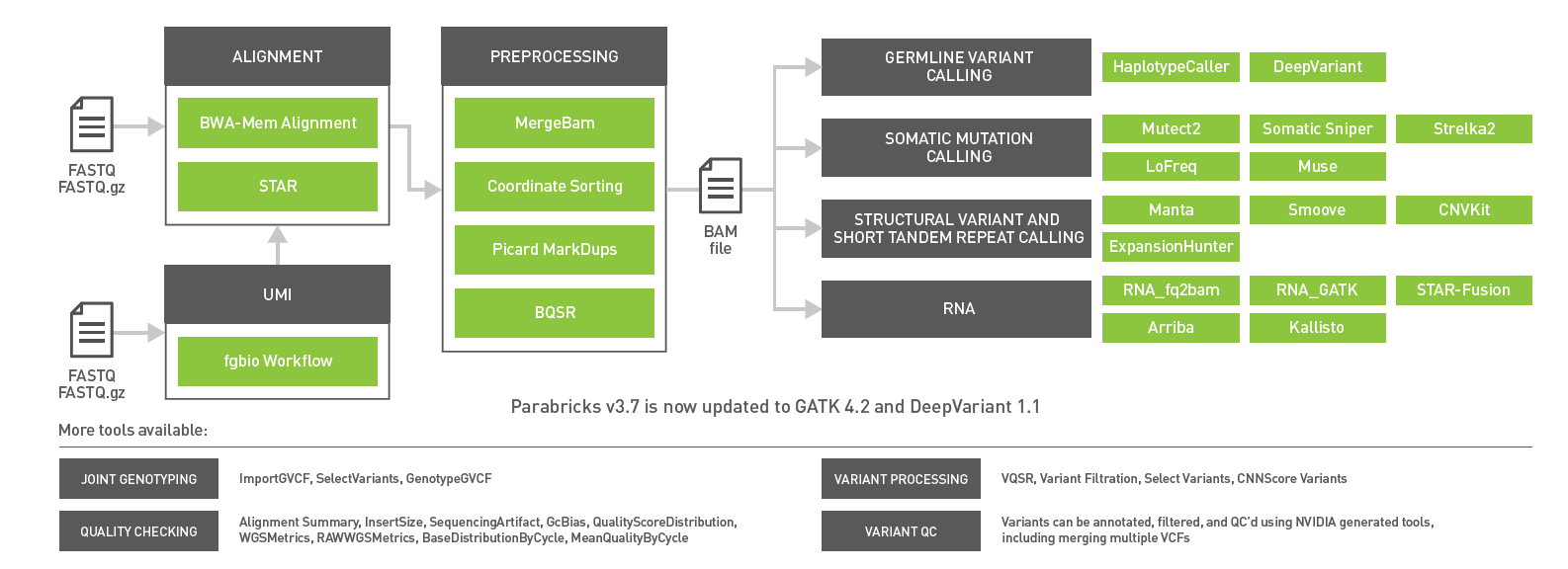
The newest release of GPU-powered NVIDIA Clara Parabricks v3.7 includes updates to germline variant callers, enhanced support for RNA-Seq pipelines, and optimized workflows for gene panels. With now over 50 tools, Clara Parabricks powers accurate and accelerated genomic analysis for gene panels, exomes, and genomes for clinical and research workflows.
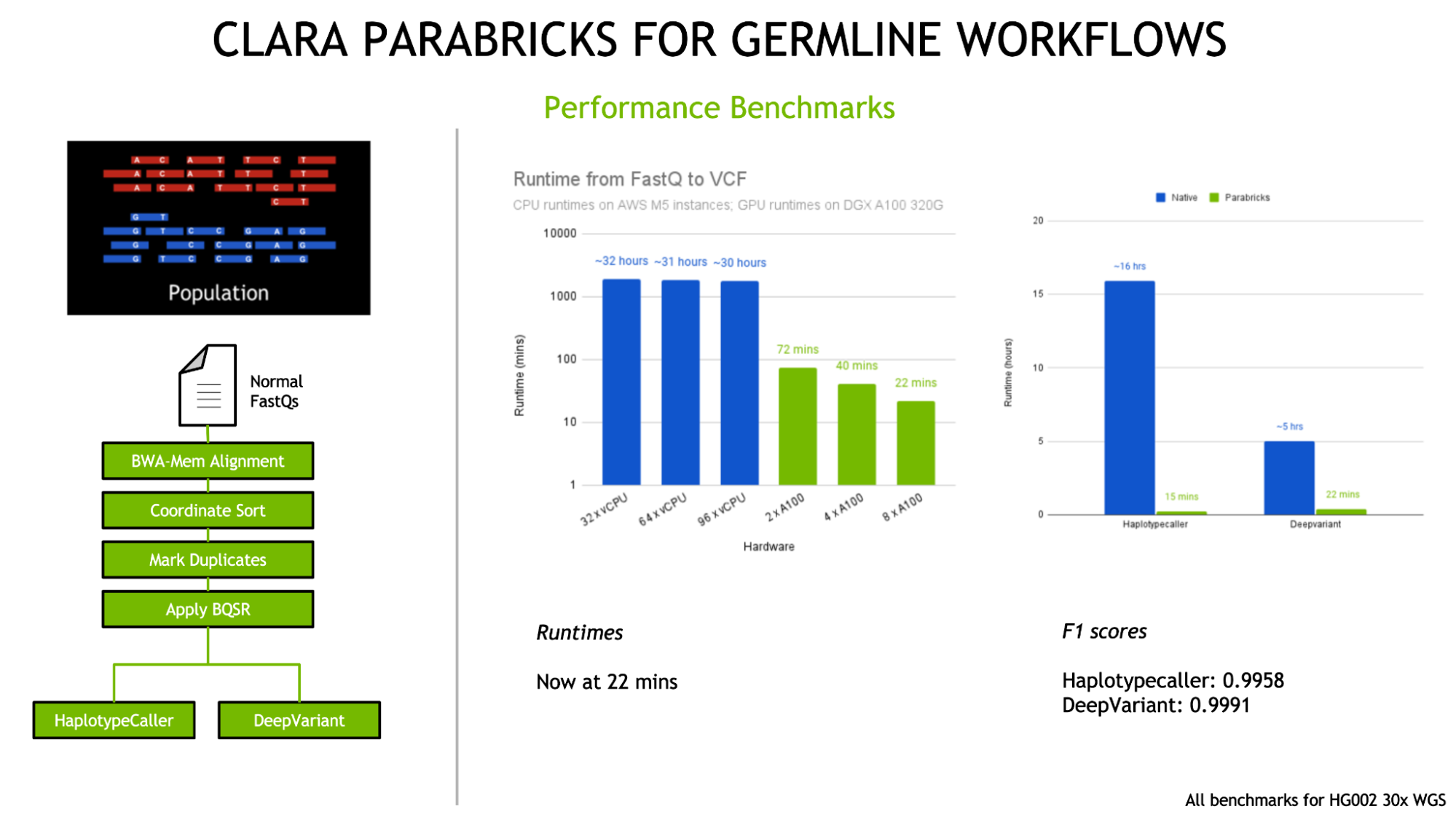
To date, Clara Parabricks has demonstrated 60x accelerations for state-of-the-art bioinformatics tools for whole genome workflows (end-to-end analysis in 22 minutes) and exome workflows (end-to-end analysis in 4 minutes), compared to CPU-based environments. Large-scale sequencing projects and other whole genome studies are able to analyze over 60 genomes/day on a single DGX server while both reducing the associated costs and generating more useful insights than ever before.
Many organizations including The National BioBank of Thailand, Human Genome Center and University of Tokyo, Translational Genomics Research Institute, Washington University in St. Louis, Regeneron Genetics Center and the UK Biobank, and Euan Ashley’s lab at Stanford University are using Clara Parabricks for fast genome and exome analysis for large population projects, critically-ill patients, and other cancer and inherited disease projects. Their work aims to accurately and quickly identify disease-causing variants, keeping pace with accelerated next-generation sequencing as well as accelerated genomic analyses.
NVIDIA Clara Parabricks v3.7 overview
- Accelerate and simplify gene panel workflows with the support of Unique Molecular Identifiers (UMIs) also known as molecular barcodes.
- RNA-Seq support for transcriptome workflows with second gene fusion caller Arriba and RNA-Seq quantification tool Kallisto.
- Short tandem repeat (STR) detection with ExpansionHunter.
- Integration of the latest versions of germline callers DeepVariant v1.1 and GATK v4.2 with HaplotypeCaller.
- A 10x accelerated BAM2FASTQ tool for converting archived data stored as either BAM or CRAM files back to FASTQ. Datasets can be updated by aligning to new and improved references.
Clara Parabricks 3.7 accelerates and simplifies gene panel analysis
While whole genome sequencing (WGS) is growing due to large-scale population initiatives, gene panels still dominate clinical genomic analysis. With time being one of the most important factors in clinical care, accelerating and simplifying gene-panel workflows is incredibly important for clinical sequencing centers. By further reducing the analysis bottleneck associated with gene panels, these sequencing centers can return results to clinicians faster, improving the quality of life for their patients.
Cancer samples used for gene panels are commonly derived from either a solid tumor or consist of cell-free DNA from the blood (liquid biopsies) of a patient. Compared to the discovery work in WGS, gene panels are narrowly focused on identifying genetic variants in known genes that either cause disease or can be targeted with specific therapies.
Gene panels for inherited diseases are often sequenced to 100x coverage while gene panels in cancer sequencing are sequenced to a much higher depth, up to several 1,000x for liquid biopsy samples. The higher coverage is required to detect lower frequency somatic mutations associated with cancer.
To improve the limit of detection for these gene panels, molecular barcodes or UMIs are used, as they significantly reduce the background noise. This limit of detection is pushed for liquid biopsies, and can include tens of thousands coverage in combination with UMIs to identify those needle-in-the-haystack somatic mutations circulating in the bloodstream. High-depth gene-panel sequencing can reintroduce a computational bottleneck in the required processing of many more sequencing reads.
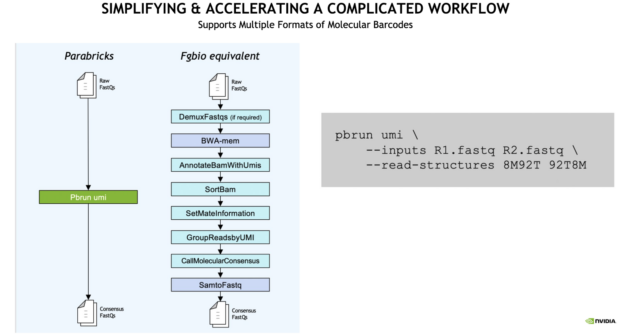
With Clara Parabricks, UMI gene panels can now be processed 10x faster than traditional workflows, generating results in less than an hour.
The analysis workflow is also simplified. From raw FASTQ to consensus FASTQ, a single command line runs multiple inputs compared to the traditional Fulcrum Genomics (Fgbio) equivalent as seen in the example below.
RNA-Seq support with Arriba and Kallisto
Just as gene panels are important for sequencing cancer analysis, so too are RNA-Seq workflows for transcriptome analysis. In addition to STAR-fusion, Clara Parabricks v3.7 now includes Arriba, a fusion detection algorithm based on the STARRNA-Seq aligner. Gene fusions, in which two distinct genes join due to a large chromosomal alteration, are associated with many different types of cancer from leukemia to solid tumors.
Arriba can also detect viral integration sites, internal tandem duplications, whole exon duplications, circular RNAs, enhancer hijacking events involving immunoglobulin/T-cell receptor loci, and breakpoints in introns or intergenic regions.
Clara Parabricks v3.7 also incorporates Kallisto, a fast RNA-Seq quantification tool based on pseudo-alignment that identifies transcript abundances (aka gene expression levels based on sequencing read counts) from either bulk or single-cell RNA-Seq datasets. Alignment of RNA-Seq data is the first step of the RNA-Seq analysis workflow. With tools for transcript quantification, read alignment, and fusion calling, Clara Parabricks 3.7 now provides a full suite of tools to support multiple RNA-Seq workflows.
Short tandem repeat detection with ExpansionHunter
To support genotyping of short tandem repeats (STRs) from short-read sequencing data, ExpansionHunter support has been added to Parabricks v3.7. STRs, also referred to as microsatellites, are ubiquitous in the human genome. These regions of noncoding DNA have accordion-like stretches of DNA containing core repeat units between two and seven nucleotides in length, repeated in tandem, up to several dozen times.
STRs are extremely useful in applications such as the construction of genetic maps, gene location, genetic linkage analysis, identification of individuals, paternity testing, population genetics, and disease diagnosis.
There are a number of regions in the human genome consisting of such repeats, which can expand in their number of repetitions, causing disease. Fragile X Syndrome, ALS, and Huntington’s Disease are well-known examples of repeat-associated diseases.
ExpansionHunter aims to estimate sizes of repeats by performing a targeted search through a BAM/CRAM file for sequencing reads that span, flank, and are fully contained in each STR.
The addition of somatic callers and support of archived data
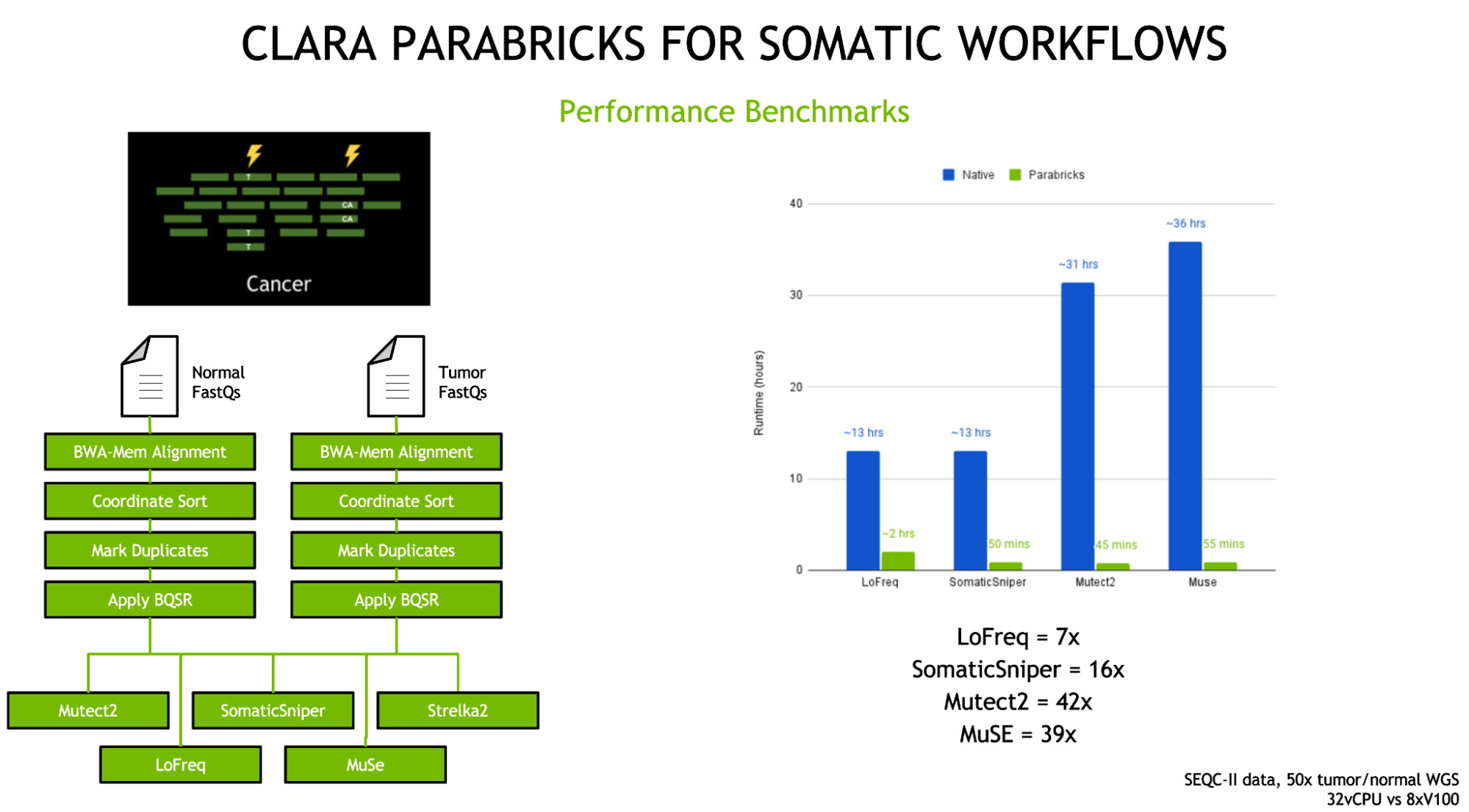
The previous releases of Clara Parabricks in 2021 brought a host of new tools, most importantly the addition of five somatics callers—MuSE, LoFreq, Strelka2, Mutect2, and SomaticSniper—for comprehensive cancer genomic analysis.
In addition, tools were added to take advantage of archived data in scenarios when original FASTQ files were deleted to save storage space. BAM2FASTQ is an accelerated version of GATK Sam2fastq, which converts an existing BAM or CRAM file to a FASTQ file. This allows users to realign sequencing reads to a new reference genome which will enable more variants to be called, along with providing researchers capabilities to normalize all their data to the same reference. Using the existing fq2bam tool, Clara Parabricks can realign reads from one reference to another in under 2 hours for a 30X whole genome.
- Try out GPU-accelerated NVIDIA Clara Parabricks genomic analysis tools for your germline, cancer, and RNA-Seq analysis workflows with a free with a 90-day evaluation trial license.
- Access NVIDIA Clara Parabricks on premise or in the cloud through the AWS Marketplace.
- Read more about Parabricks v3.7 release.