With a new year underway, NVIDIA is helping enterprises worldwide add modern workloads to their mainstream servers using the latest release of the NVIDIA AI Enterprise software suite. NVIDIA AI Enterprise 1.1 is now generally available. Optimized, certified and supported by NVIDIA, the latest version of the software suite brings new updates including production support Read article >
How much would it take to train a model which consists of about 2000 pictures on my laptop (I am a beginner but I need to train it for a project) Specs: Ryzen 5 3500u 10gb of ram vega 8 gpu
I was going to do it on the cpu, because I think its more powerful (correct me if I am wrong)
Posted by Oran Lang and Inbar Mosseri, Software Engineers, Google Research
Neural networks can perform certain tasks remarkably well, but understanding how they reach their decisions — e.g., identifying which signals in an image cause a model to determine it to be of one class and not another — is often a mystery. Explaining a neural model’s decision process may have high social impact in certain areas, such as analysis of medical images and autonomous driving, where human oversight is critical. These insights can also be helpful in guiding health care providers, revealing model biases, providing support for downstream decision makers, and even aiding scientific discovery.
Previous approaches for visual explanations of classifiers, such as attention maps (e.g., Grad-CAM), highlight which regions in an image affect the classification, but they do not explain what attributes within those regions determine the classification outcome: For example, is it their color? Their shape? Another family of methods provides an explanation by smoothly transforming the image between one class and another (e.g., GANalyze). However, these methods tend to change all attributes at once, thus making it difficult to isolate the individual affecting attributes.
In “Explaining in Style: Training a GAN to explain a classifier in StyleSpace”, presented at ICCV 2021, we propose a new approach for a visual explanation of classifiers. Our approach, StylEx, automatically discovers and visualizes disentangled attributes that affect a classifier. It allows exploring the effect of individual attributes by manipulating those attributes separately (changing one attribute does not affect others). StylEx is applicable to a wide range of domains, including animals, leaves, faces, and retinal images. Our results show that StylEx finds attributes that align well with semantic ones, generate meaningful image-specific explanations, and are interpretable by people as measured in user studies.
Explaining a Cat vs. Dog Classifier: StylEx provides the top-K discovered disentangled attributes which explain the classification. Moving each knob manipulates only the corresponding attribute in the image, keeping other attributes of the subject fixed.
For instance, to understand a cat vs. dog classifier on a given image, StylEx can automatically detect disentangled attributes and visualize how manipulating each attribute can affect the classifier probability. The user can then view these attributes and make semantic interpretations for what they represent. For example, in the figure above, one can draw conclusions such as “dogs are more likely to have their mouth open than cats” (attribute #4 in the GIF above), “cats’ pupils are more slit-like” (attribute #5), “cats’ ears do not tend to be folded” (attribute #1), and so on.
The video below provides a short explanation of the method:
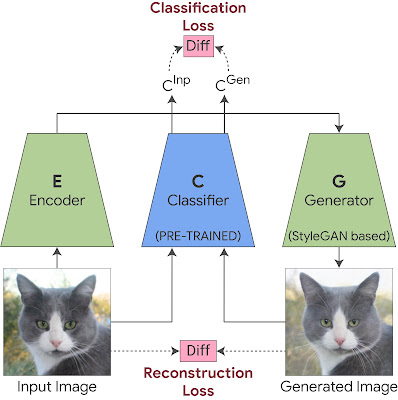
How StylEx Works: Training StyleGAN to Explain a Classifier Given a classifier and an input image, we want to find and visualize the individual attributes that affect its classification. For that, we utilize the StyleGAN2 architecture, which is known to generate high quality images. Our method consists of two phases:
Phase 1: Training StylEx
A recent work showed that StyleGAN2 contains a disentangled latent space called “StyleSpace”, which contains individual semantically meaningful attributes of the images in the training dataset. However, because StyleGAN training is not dependent on the classifier, it may not represent those attributes that are important for the decision of the specific classifier we want to explain. Therefore, we train a StyleGAN-like generator to satisfy the classifier, thus encouraging its StyleSpace to accommodate classifier-specific attributes.
This is achieved by training the StyleGAN generator with two additional components. The first is an encoder, trained together with the GAN with a reconstruction-loss, which forces the generated output image to be visually similar to the input. This allows us to apply the generator on any given input image. However, visual similarity of the image is not enough, as it may not necessarily capture subtle visual details important for a particular classifier (such as medical pathologies). To ensure this, we add a classification-loss to the StyleGAN training, which forces the classifier probability of the generated image to be the same as the classifier probability of the input image. This guarantees that subtle visual details important for the classifier (such as medical pathologies) will be included in the generated image.
Training StyleEx: We jointly train the generator and the encoder. A reconstruction-loss is applied between the generated image and the original image to preserve visual similarity. A classification-loss is applied between the classifier output of the generated image and the classifier output of the original image to ensure the generator captures subtle visual details important for the classification.
Phase 2: Extracting Disentangled Attributes
Once trained, we search the StyleSpace of the trained Generator for attributes that significantly affect the classifier. To do so, we manipulate each StyleSpace coordinate and measure its effect on the classification probability. We seek the top attributes that maximize the change in classification probability for the given image. This provides the top-K image-specific attributes. By repeating this process for a large number of images per class, we can further discover the top-K class-specific attributes, which teaches us what the classifier has learned about the specific class. We call our end-to-end system “StylEx”.
A visual illustration of image-specific attribute extraction: once trained, we search for the StyleSpace coordinates that have the highest effect on the classification probability of a given image.
StylEx is Applicable to a Wide Range of Domains and Classifiers Our method works on a wide variety of domains and classifiers (binary and multi-class). Below are some examples of class-specific explanations. In all the domains tested, the top attributes detected by our method correspond to coherent semantic notions when interpreted by humans, as verified by human evaluation.
For perceived gender and age classifiers, below are the top four detected attributes per classifier. Our method exemplifies each attribute on multiple images that are automatically selected to best demonstrate that attribute. For each attribute we flicker between the source and attribute-manipulated image. The degree to which manipulating the attribute affects the classifier probability is shown at the top-left corner of each image.
Top-4 automatically detected attributes for a perceived-gender classifier.
Top-4 automatically detected attributes for a perceived-age classifier.
Note that our method explains a classifier, not reality. That is, the method is designed to reveal image attributes that a given classifier has learned to utilize from data; those attributes may not necessarily characterize actual physical differences between class labels (e.g., a younger or older age) in reality. In particular, these detected attributes may reveal biases in the classifier training or dataset, which is another key benefit of our method. It can further be used to improve fairness of neural networks, for example, by augmenting the training dataset with examples that compensate for the biases our method reveals.
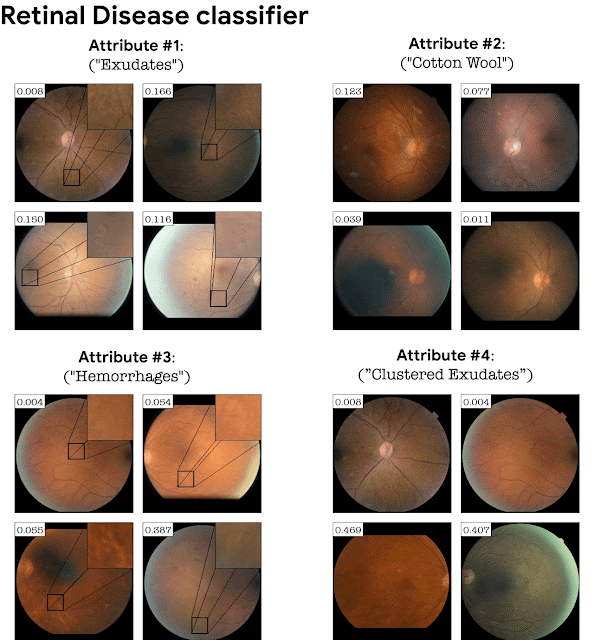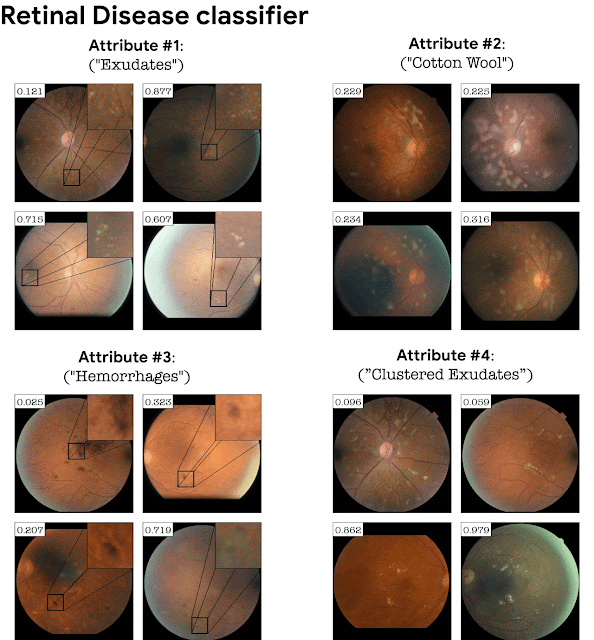
Adding the classifier loss into StyleGAN training turns out to be crucial in domains where the classification depends on fine details. For example, a GAN trained on retinal images without a classifier loss will not necessarily generate fine pathological details corresponding to a particular disease. Adding the classification loss causes the GAN to generate these subtle pathologies as an explanation of the classifier. This is exemplified below for a retinal image classifier (DME disease) and a sick/healthy leaf classifier. StylEx is able to discover attributes that are aligned with disease indicators, for instance “hard exudates”, which is a well known marker for retinal DME, and rot for leaf diseases.
Top-4 automatically detected attributes for a DME classifier of retina images.
Top-4 automatically detected attributes for a classifier of sick/healthy leaf images.
Finally, this method is also applicable to multi-class problems, as demonstrated on a 200-way bird species classifier.
Top-4 automatically detected attributes in a 200-way classifier trained on CUB-2011 for(a) the class “brewer blackbird”, and (b) the class “yellow bellied flycatcher”. Indeed we observe that StylEx detects attributes that correspond to attributes in CUB taxonomy.
Broader Impact and Next Steps Overall, we have introduced a new technique that enables the generation of meaningful explanations for a given classifier on a given image or class. We believe that our technique is a promising step towards detection and mitigation of previously unknown biases in classifiers and/or datasets, in line with Google’s AI Principles. Additionally, our focus on multiple-attribute based explanation is key to providing new insights about previously opaque classification processes and aiding in the process of scientific discovery. Finally, our GitHub repository includes a Colab and model weights for the GANs used in our paper.
Acknowledgements The research described in this post was done by Oran Lang, Yossi Gandelsman, Michal Yarom, Yoav Wald (as an intern), Gal Elidan, Avinatan Hassidim, William T. Freeman, Phillip Isola, Amir Globerson, Michal Irani and Inbar Mosseri. We would like to thank Jenny Huang and Marilyn Zhang for leading the writing process for this blogpost, and Reena Jana, Paul Nicholas, and Johnny Soraker for ethics reviews of our research paper and this post.
GANcraft is a hybrid neural rendering pipeline to represent large and complex scenes using Minecraft.
Scientists at NVIDIA and Cornell University introduced a hybrid unsupervised neural rendering pipeline to represent large and complex scenes efficiently in voxel worlds. Essentially, a 3D artist only needs to build the bare minimum, and the algorithm will do the rest to build a photorealistic world. The researchers applied this hybrid neural rendering pipeline to Minecraft block worlds to generate a far more realistic version of the Minecraft scenery.
Previous works from NVIDIA and the broader research community (pix2pix, pix2pixHD, MUNIT, SPADE) have tackled the problem of image-to-image translation (im2im)—translating an image from one domain to another. At first glance, these methods might seem to offer a simple solution to the task of transforming one world to another—translating one image at a time. However, im2im methods do not preserve viewpoint consistency, as they have no knowledge of the 3D geometry, and each 2D frame is generated independently. As can be seen in the images that follow, the results from these methods produce jitter and abrupt color and texture changes.
MUNIT SPADE wc-vid2vid NSVF-W GANcraft
Figure 1. A comparison of prior works and GANcraft.
Enter GANcraft, a new method that directly operates on the 3D input world.
“As the ground truth photorealistic renderings for a user-created block world simply doesn’t exist, we have to train models with indirect supervision,” the researchers explained in the study.
The method works by randomly sampling camera views in the input block world and then imagining what a photorealistic version of that view would look like. This is done with the help of SPADE, prior work from NVIDIA on image-to-image translation, and was the key component in the popular GauGAN demo. GANcraft overcomes the view inconsistency of these generated “pseudo-groundtruths” through the use of a style-conditioning network that can disambiguate the world structure from the rendering style. This enables GANcraft to generate output videos that are view consistent, as well as with different styles as shown in this image!
Figure 2. GANcraft’s methodology enables view consistency in a variety of different styles.
While the results of the research are demonstrated in Minecraft, the method works with other 3D block worlds such as voxels. The potential to shorten the amount of time and expertise needed to build high-definition worlds increases the value of this research. It could help game developers, CGI artists, and the animation industry cut down on the time it takes to build these large and impressive worlds.
If you would like a further breakdown of the potential of this technology, Károly Zsolnai-Fehér highlights the research in his YouTube series: Two Minute Papers:
Figure 3. The YouTube series, Two Minute Papers, covers significant developments in AI as they come onto the scene.
GANcraft was implemented in the Imaginaire library. This library is optimized for the training of generative models and generative adversarial networks, with support for multi-GPU, multi-node, and automatic mixed-precision training. Implementations of over 10 different research works produced by NVIDIA, as well as pretrained models have been released. This library will continue to be updated with newer works over time.
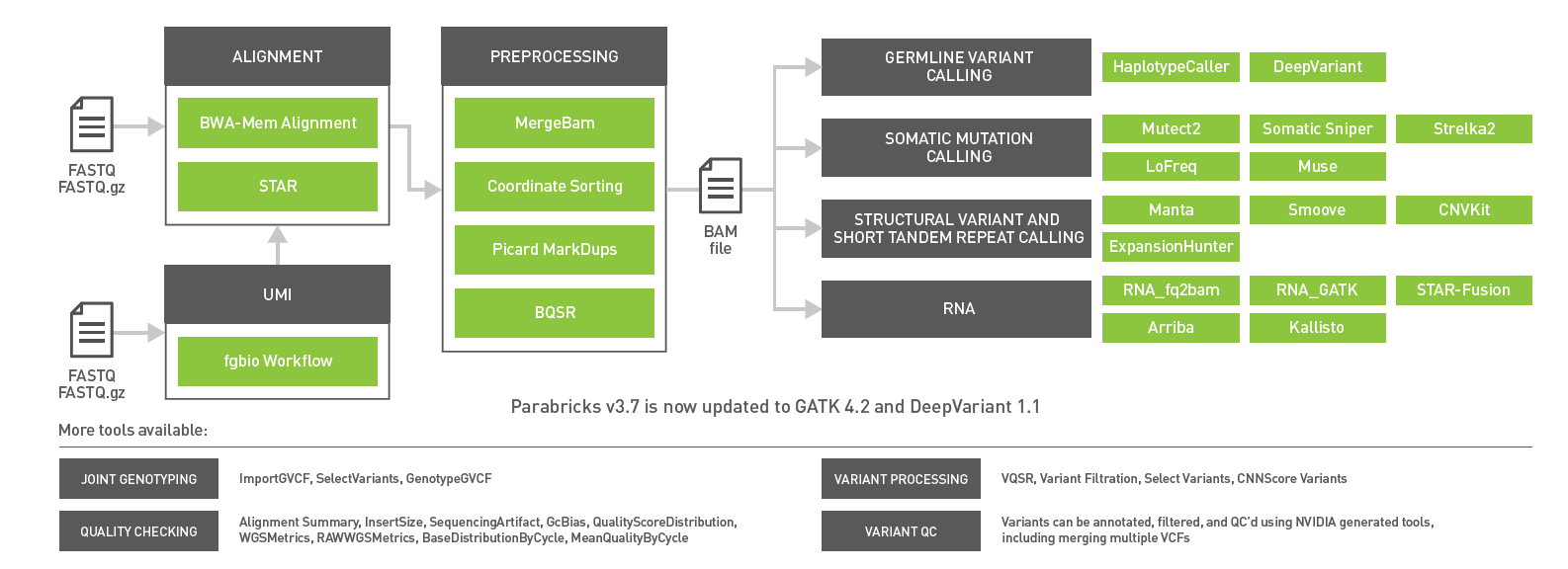
The GPU-accelerated Clara Parabricks v3.7 release brings support for gene panels, RNA-Seq, short tandem repeats, and updates to GATK 4.2 and DeepVariant 1.1.
The newest release of GPU-powered NVIDIA Clara Parabricks v3.7 includes updates to germline variant callers, enhanced support for RNA-Seq pipelines, and optimized workflows for gene panels. With now over 50 tools, Clara Parabricks powers accurate and accelerated genomic analysis for gene panels, exomes, and genomes for clinical and research workflows.
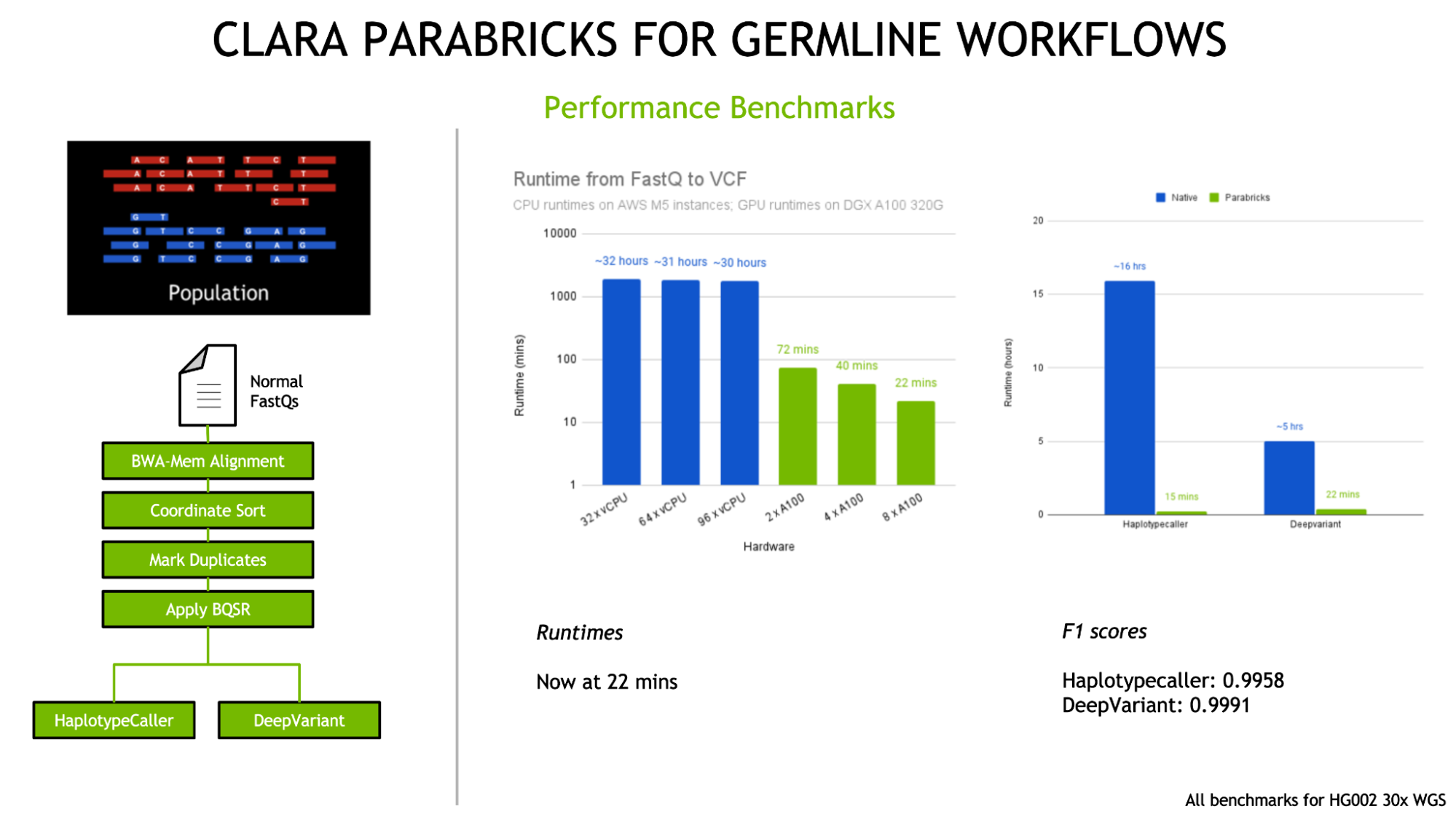
To date, Clara Parabricks has demonstrated 60x accelerations for state-of-the-art bioinformatics tools for whole genome workflows (end-to-end analysis in 22 minutes) and exome workflows (end-to-end analysis in 4 minutes), compared to CPU-based environments. Large-scale sequencing projects and other whole genome studies are able to analyze over 60 genomes/day on a single DGX server while both reducing the associated costs and generating more useful insights than ever before.
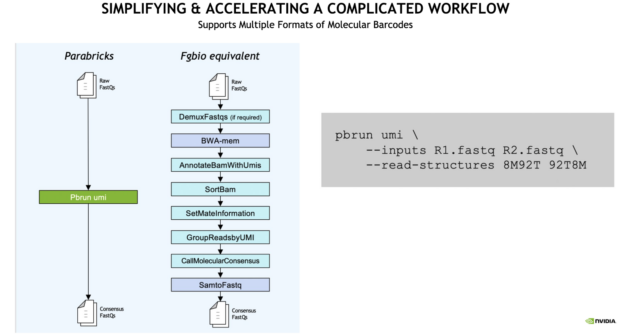
Accelerate and simplify gene panel workflows with the support of Unique Molecular Identifiers (UMIs) also known as molecular barcodes.
RNA-Seq support for transcriptome workflows with second gene fusion caller Arriba and RNA-Seq quantification tool Kallisto.
Short tandem repeat (STR) detection with ExpansionHunter.
Integration of the latest versions of germline callers DeepVariant v1.1 and GATK v4.2 with HaplotypeCaller.
A 10x accelerated BAM2FASTQ tool for converting archived data stored as either BAM or CRAM files back to FASTQ. Datasets can be updated by aligning to new and improved references.
Figure 1: Clara Parabricks 3.7 includes a UMI workflow for gene panels, ExpansionHunter for short tandem repeats, inclusion of the DeepVariant 1.1 and GATK 4.2 releases, and Kallisto and Arriba for RNA-Seq workflows.
Clara Parabricks 3.7 accelerates and simplifies gene panel analysis
While whole genome sequencing (WGS) is growing due to large-scale population initiatives, gene panels still dominate clinical genomic analysis. With time being one of the most important factors in clinical care, accelerating and simplifying gene-panel workflows is incredibly important for clinical sequencing centers. By further reducing the analysis bottleneck associated with gene panels, these sequencing centers can return results to clinicians faster, improving the quality of life for their patients.
Cancer samples used for gene panels are commonly derived from either a solid tumor or consist of cell-free DNA from the blood (liquid biopsies) of a patient. Compared to the discovery work in WGS, gene panels are narrowly focused on identifying genetic variants in known genes that either cause disease or can be targeted with specific therapies.
Gene panels for inherited diseases are often sequenced to 100x coverage while gene panels in cancer sequencing are sequenced to a much higher depth, up to several 1,000x for liquid biopsy samples. The higher coverage is required to detect lower frequency somatic mutations associated with cancer.
To improve the limit of detection for these gene panels, molecular barcodes or UMIs are used, as they significantly reduce the background noise. This limit of detection is pushed for liquid biopsies, and can include tens of thousands coverage in combination with UMIs to identify those needle-in-the-haystack somatic mutations circulating in the bloodstream. High-depth gene-panel sequencing can reintroduce a computational bottleneck in the required processing of many more sequencing reads.
With Clara Parabricks, UMI gene panels can now be processed 10x faster than traditional workflows, generating results in less than an hour.
The analysis workflow is also simplified. From raw FASTQ to consensus FASTQ, a single command line runs multiple inputs compared to the traditional Fulcrum Genomics (Fgbio) equivalent as seen in the example below.
Figure 2: Clara Parabricks 3.7 has simplified and accelerated gene panel UMI workflows compared to the Fgbio equivalent.
RNA-Seq support with Arriba and Kallisto
Just as gene panels are important for sequencing cancer analysis, so too are RNA-Seq workflows for transcriptome analysis. In addition to STAR-fusion, Clara Parabricks v3.7 now includes Arriba, a fusion detection algorithm based on the STARRNA-Seq aligner. Gene fusions, in which two distinct genes join due to a large chromosomal alteration, are associated with many different types of cancer from leukemia to solid tumors.
Arriba can also detect viral integration sites, internal tandem duplications, whole exon duplications, circular RNAs, enhancer hijacking events involving immunoglobulin/T-cell receptor loci, and breakpoints in introns or intergenic regions.
Clara Parabricks v3.7 also incorporates Kallisto, a fast RNA-Seq quantification tool based on pseudo-alignment that identifies transcript abundances (aka gene expression levels based on sequencing read counts) from either bulk or single-cell RNA-Seq datasets. Alignment of RNA-Seq data is the first step of the RNA-Seq analysis workflow. With tools for transcript quantification, read alignment, and fusion calling, Clara Parabricks 3.7 now provides a full suite of tools to support multiple RNA-Seq workflows.
Short tandem repeat detection with ExpansionHunter
To support genotyping of short tandem repeats (STRs) from short-read sequencing data, ExpansionHunter support has been added to Parabricks v3.7. STRs, also referred to as microsatellites, are ubiquitous in the human genome. These regions of noncoding DNA have accordion-like stretches of DNA containing core repeat units between two and seven nucleotides in length, repeated in tandem, up to several dozen times.
STRs are extremely useful in applications such as the construction of genetic maps, gene location, genetic linkage analysis, identification of individuals, paternity testing, population genetics, and disease diagnosis.
There are a number of regions in the human genome consisting of such repeats, which can expand in their number of repetitions, causing disease. Fragile X Syndrome, ALS, and Huntington’s Disease are well-known examples of repeat-associated diseases.
ExpansionHunter aims to estimate sizes of repeats by performing a targeted search through a BAM/CRAM file for sequencing reads that span, flank, and are fully contained in each STR.
The addition of somatic callers and support of archived data
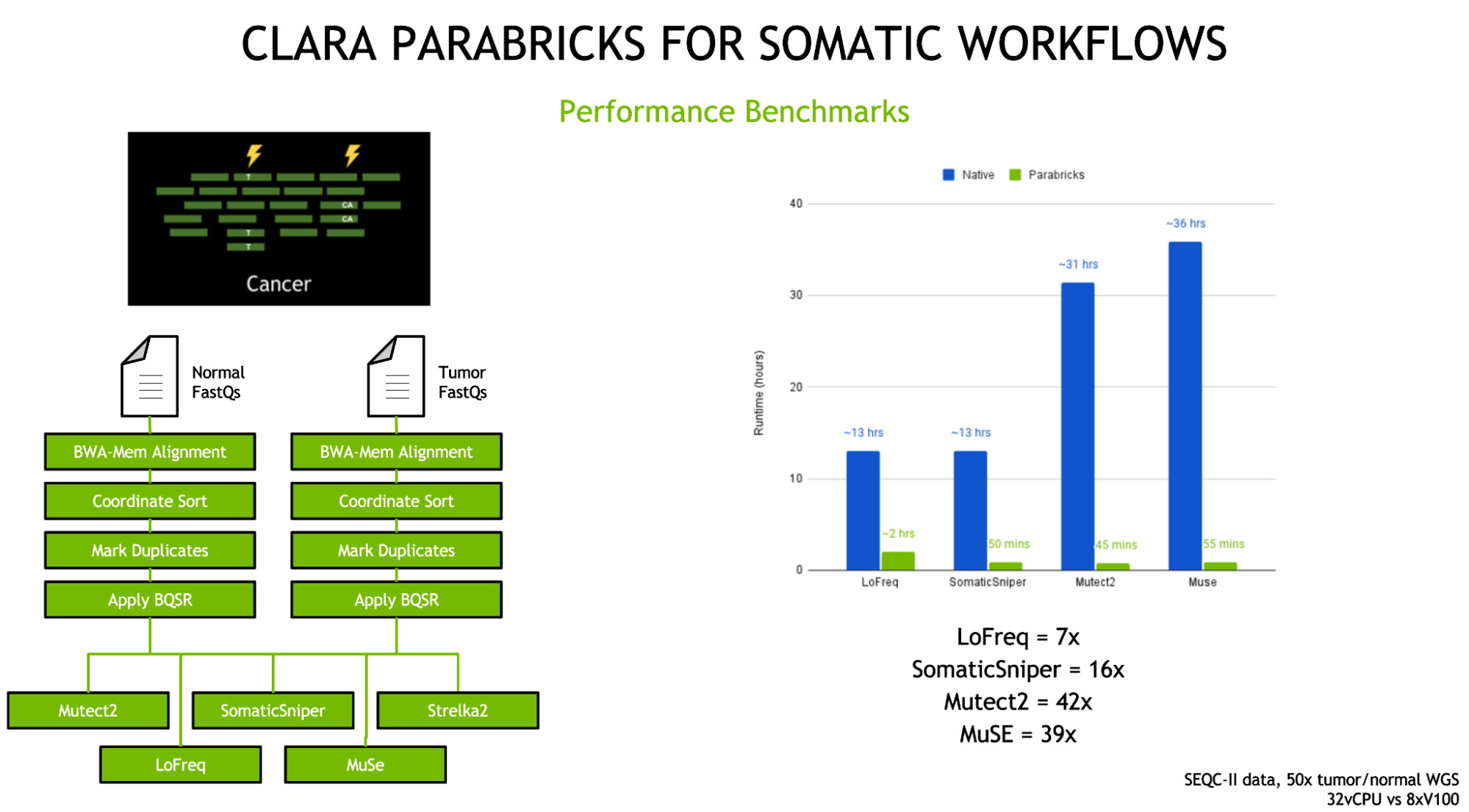
The previous releases of Clara Parabricks in 2021 brought a host of new tools, most importantly the addition of five somatics callers—MuSE, LoFreq, Strelka2, Mutect2, and SomaticSniper—for comprehensive cancer genomic analysis.
Figure 3: Clara Parabricks v3.6.1 and 3.7 includes five somatic callers for comprehensive accelerated cancer genomic analysis—Muse, LoFreq, Strelka2, Mutect2, and SomaticSniper.
In addition, tools were added to take advantage of archived data in scenarios when original FASTQ files were deleted to save storage space. BAM2FASTQ is an accelerated version of GATK Sam2fastq, which converts an existing BAM or CRAM file to a FASTQ file. This allows users to realign sequencing reads to a new reference genome which will enable more variants to be called, along with providing researchers capabilities to normalize all their data to the same reference. Using the existing fq2bam tool, Clara Parabricks can realign reads from one reference to another in under 2 hours for a 30X whole genome.
Figure 4: Clara Parabricks v3.7 has added the latest releases of germline variant callers DeepVariant 1.1 and GATK 4.2 with HaplotypeCaller.
Try out GPU-accelerated NVIDIA Clara Parabricks genomic analysis tools for your germline, cancer, and RNA-Seq analysis workflows with a free with a 90-day evaluation trial license.
Online commerce has rocketed to trillions of dollars worldwide in the past decade, serving billions of consumers. Behind the scenes of this explosive growth in online sales is personalization driven by recommender engines. Recommenders make shopping deeply personalized. While searching for products on e-commerce sites, they find you. Or suggestions can just appear. This wildly Read article >
Hello, I’m currently working through the tensorflow agent and bandit library/tutorials. But I can’t answer completely myself what the difference between the driver and the trainer is. Is it basically the trainer is training the agent and for example his neuronal network for the (approximately) perfect policy in a given environment? And the the driver is just the execution of a given policy without any regards to optimizing it along the way ?
Does anyone know if it’s possible to compile tensorflow lite (C api) as a static library, and more specifically for iOS?
I have a C library that needs to interact with tensorflow lite, which I want to integrate into an iOS application, but iOS only accepts static libraries
Didn’t see that the imgDimentions on the trainer was set to 225,225,1 but the detector was trying to find 255,255,1 images
its been a long day!
Hi all, me again haha
Ive run into an error that has stumped me for about an hour;
Im trying to train a TensorFlow modal to detect a British one pence coin and a British 2 pence coin, while the model trained, when it detects the objects it crashes and gives me this error:
ValueError: Input 0 of layer “sequential” is incompatible with the layer: expected shape=(None, 225, 225, 1), found shape=(None, 255, 255, 1)
its finding the shape its supposed to find, but it says its not? Any advice would be greatly appreciated!
I recently started doing some smaller TF projects and decided that it’s a good time to get some more formal training in the area and with TF.
I found a few interesting resources and was wondering if any of you have completed them and what your thought are? Are there any great resources I am missing on this list?
For background, I am most interested in time series analysis, medical AI, and TF on embedded devices like the Google Coral but think a combination of a broad course + one specialized on these areas would be best.








 GANcraft is a hybrid neural rendering pipeline to represent large and complex scenes using Minecraft.
GANcraft is a hybrid neural rendering pipeline to represent large and complex scenes using Minecraft.




 The GPU-accelerated Clara Parabricks v3.7 release brings support for gene panels, RNA-Seq, short tandem repeats, and updates to GATK 4.2 and DeepVariant 1.1.
The GPU-accelerated Clara Parabricks v3.7 release brings support for gene panels, RNA-Seq, short tandem repeats, and updates to GATK 4.2 and DeepVariant 1.1.