
 The release of NVIDIA Clara Parabricks v3.6 brings new applications for variant calling, annotation, filtering, and quality control to its suite of powerful genomic analysis tools. Now featuring over 33 accelerated tools for every stage of genomic analysis, NVIDIA Clara Parabricks provides GPU-accelerated bioinformatic pipelines that can scale for any workload. As genomes and exomes … Continued
The release of NVIDIA Clara Parabricks v3.6 brings new applications for variant calling, annotation, filtering, and quality control to its suite of powerful genomic analysis tools. Now featuring over 33 accelerated tools for every stage of genomic analysis, NVIDIA Clara Parabricks provides GPU-accelerated bioinformatic pipelines that can scale for any workload. As genomes and exomes … Continued
The release of NVIDIA Clara Parabricks v3.6 brings new applications for variant calling, annotation, filtering, and quality control to its suite of powerful genomic analysis tools. Now featuring over 33 accelerated tools for every stage of genomic analysis, NVIDIA Clara Parabricks provides GPU-accelerated bioinformatic pipelines that can scale for any workload.
As genomes and exomes are sequenced at faster speeds than ever before, increasing loads of raw instrument data must be mapped, aligned, and interpreted to decipher variants and their significance to disease. Bioinformatic pipelines need to keep up with genomic analysis tools. CPU-based analysis pipelines often take weeks or months to glean results, while GPU-based pipelines can analyze 30X whole human genomes in 22 minutes and whole human exomes in 4 minutes.
These fast turnaround times are necessary to keep pace with next generation sequencing (NGS) genomic instrument outputs. This is imperative for large-scale population, cancer center, pharmaceutical drug development, and genomic research projects that require quick results for publications.
NVIDIA Clara Parabricks v3.6 Incorporates:
- New GPU-accelerated variant callers
- An easy-to-use vote-based VCF merging tool (VBVM)
- A database annotation tool (VCFANNO)
- A new tool for quickly filtering a VCF by allele frequency (FrequencyFiltration)
- Tools for VCF quality control (VCFQC and VCFQCbyBAM) for both somatic and germline pipelines.
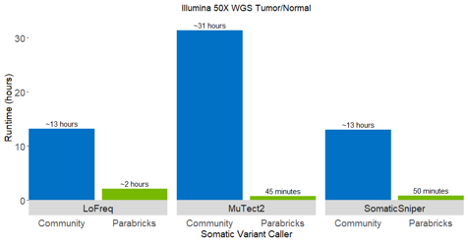
Accelerating LoFreq and Other Somatic Callers
With the addition of LoFreq alongside Strelka2, Mutect2, and SomaticSniper, Clara Parabricks now includes 4 somatic callers for cancer workflows. LoFreq is a fast and sensitive variant caller for inferring SNVs and indels from NGS data. LoFreq runs on a variety of aligned sequencing data such as Illumina, IonTorrent, and Pacbio. It can automatically adapt to changes in coverage and sequencing quality, and can be applied to somatic, viral/quasispecies, metagenomic, and bacterial datasets.
The Lofreq somatic caller in Clara Parabricks is 10X faster compared to its native instance and is ideal for calling low frequency mutations.Using base-call qualities and other sources of errors inherent in NGS data, LoFreq improves the accuracy for calling somatic mutations below the 10% allele frequency threshold.
The accelerated LoFreq supports only SNV calling in v3.6, with Indel calling coming in a subsequent release.
Read more >>
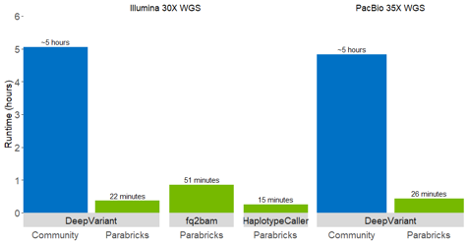
From Months to Hours with New Accelerated Tools
NVIDIA Clara Parabricks v3.6 also includes a bam2fastq tool, the addition of smoove variant callers, support for de novo mutations, and new tools for VCF processing (for example annotation, filtering, and merging). A standard WGS analysis for a 30x human genome finishes in 22 minutes on a DGX A100, which is over 80 times faster than CPU-based workflows on the same server. With this acceleration, projects taking months can now be done in hours.
Bam2Fastq is an accelerated version of GATK Sam2fastq. It converts a BAM or CRAM file to FASTQ. This is useful for scenarios where samples need to be realigned to a new reference, but the original FASTQs were deleted to save on storage space. Now they can be regenerated from the BAMs and aligned to a new reference more quickly than ever before
Detection of de novo variants (DNVs) that occur in the germline genome when comparing sequence data for an offspring to its parents (aka trio analysis) is critical for studies of disease-related variation, along with creating a baseline for generational mutation rates.
A GPU-based workflow to call DNVs is now included in NVIDIA Clara Parabricks v3.6 and utilizes Google’s DeepVariant, which has been tested on trio analyses and other pedigree sequencing projects.
Learn more >>
For structural variant calling, NVIDIA Clara Parabricks already includes Manta, and now smoove has been added. Smoove simplifies and speeds calling and genotyping structural variants for short reads. It also improves specificity by removing alignment signals indicative of low-level noise and often contribute to spurious calls.
Learn more >>
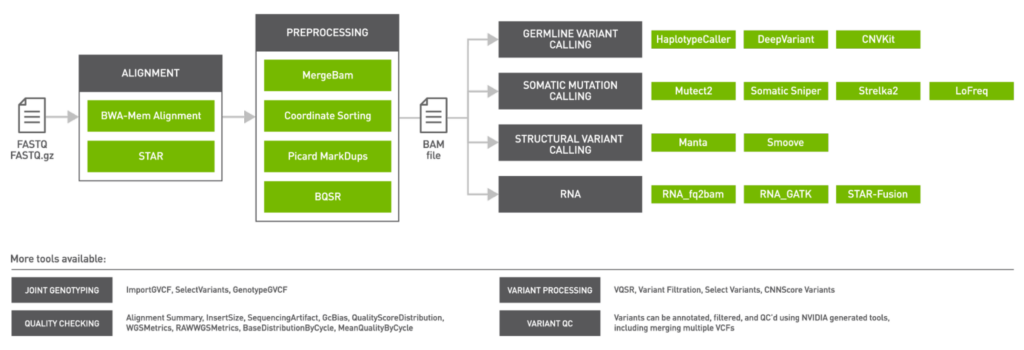
NVIDIA Clara Parabricks v3.6 also focuses on steps of the genomic pipeline after variant calling. BamBasedVCFQC is an NVIDIA-generated tool to help QC VCF outputs by using SamTools mPileUp results, using the original BAM. Vcfanno allows users to annotate VCF outputs using third-party data sources like dbSNP, adding allele frequencies to the VCF.
FrequencyFiltration allows variants within a VCF to be filtered based upon numeric fields containing allele frequency and read count information. Finally, vote-based somatic caller merger (vbvm) is for merging two or more VCF files and then filtering variants based upon a simple voting-based mechanism where variants can be filtered based upon the number of somatic callers that have identified a specific variant.
- Try out the NVIDIA Clara Parabricks GPU-accelerated genomic analysis tools for your germline, cancer, and RNA-Seq analysis workflows for free with a 90-day trial license >>>
- Access NVIDIA Clara Parabricks on premise or in the cloud through the AWS Marketplace >>>
- Read more information on v3.6 >>