GANcraft is a hybrid neural rendering pipeline to represent large and complex scenes using Minecraft.
Scientists at NVIDIA and Cornell University introduced a hybrid unsupervised neural rendering pipeline to represent large and complex scenes efficiently in voxel worlds. Essentially, a 3D artist only needs to build the bare minimum, and the algorithm will do the rest to build a photorealistic world. The researchers applied this hybrid neural rendering pipeline to Minecraft block worlds to generate a far more realistic version of the Minecraft scenery.
Previous works from NVIDIA and the broader research community (pix2pix, pix2pixHD, MUNIT, SPADE) have tackled the problem of image-to-image translation (im2im)—translating an image from one domain to another. At first glance, these methods might seem to offer a simple solution to the task of transforming one world to another—translating one image at a time. However, im2im methods do not preserve viewpoint consistency, as they have no knowledge of the 3D geometry, and each 2D frame is generated independently. As can be seen in the images that follow, the results from these methods produce jitter and abrupt color and texture changes.
MUNIT SPADE wc-vid2vid NSVF-W GANcraft
Figure 1. A comparison of prior works and GANcraft.
Enter GANcraft, a new method that directly operates on the 3D input world.
“As the ground truth photorealistic renderings for a user-created block world simply doesn’t exist, we have to train models with indirect supervision,” the researchers explained in the study.
The method works by randomly sampling camera views in the input block world and then imagining what a photorealistic version of that view would look like. This is done with the help of SPADE, prior work from NVIDIA on image-to-image translation, and was the key component in the popular GauGAN demo. GANcraft overcomes the view inconsistency of these generated “pseudo-groundtruths” through the use of a style-conditioning network that can disambiguate the world structure from the rendering style. This enables GANcraft to generate output videos that are view consistent, as well as with different styles as shown in this image!
Figure 2. GANcraft’s methodology enables view consistency in a variety of different styles.
While the results of the research are demonstrated in Minecraft, the method works with other 3D block worlds such as voxels. The potential to shorten the amount of time and expertise needed to build high-definition worlds increases the value of this research. It could help game developers, CGI artists, and the animation industry cut down on the time it takes to build these large and impressive worlds.
If you would like a further breakdown of the potential of this technology, Károly Zsolnai-Fehér highlights the research in his YouTube series: Two Minute Papers:
Figure 3. The YouTube series, Two Minute Papers, covers significant developments in AI as they come onto the scene.
GANcraft was implemented in the Imaginaire library. This library is optimized for the training of generative models and generative adversarial networks, with support for multi-GPU, multi-node, and automatic mixed-precision training. Implementations of over 10 different research works produced by NVIDIA, as well as pretrained models have been released. This library will continue to be updated with newer works over time.
The GPU-accelerated Clara Parabricks v3.7 release brings support for gene panels, RNA-Seq, short tandem repeats, and updates to GATK 4.2 and DeepVariant 1.1.
The newest release of GPU-powered NVIDIA Clara Parabricks v3.7 includes updates to germline variant callers, enhanced support for RNA-Seq pipelines, and optimized workflows for gene panels. With now over 50 tools, Clara Parabricks powers accurate and accelerated genomic analysis for gene panels, exomes, and genomes for clinical and research workflows.
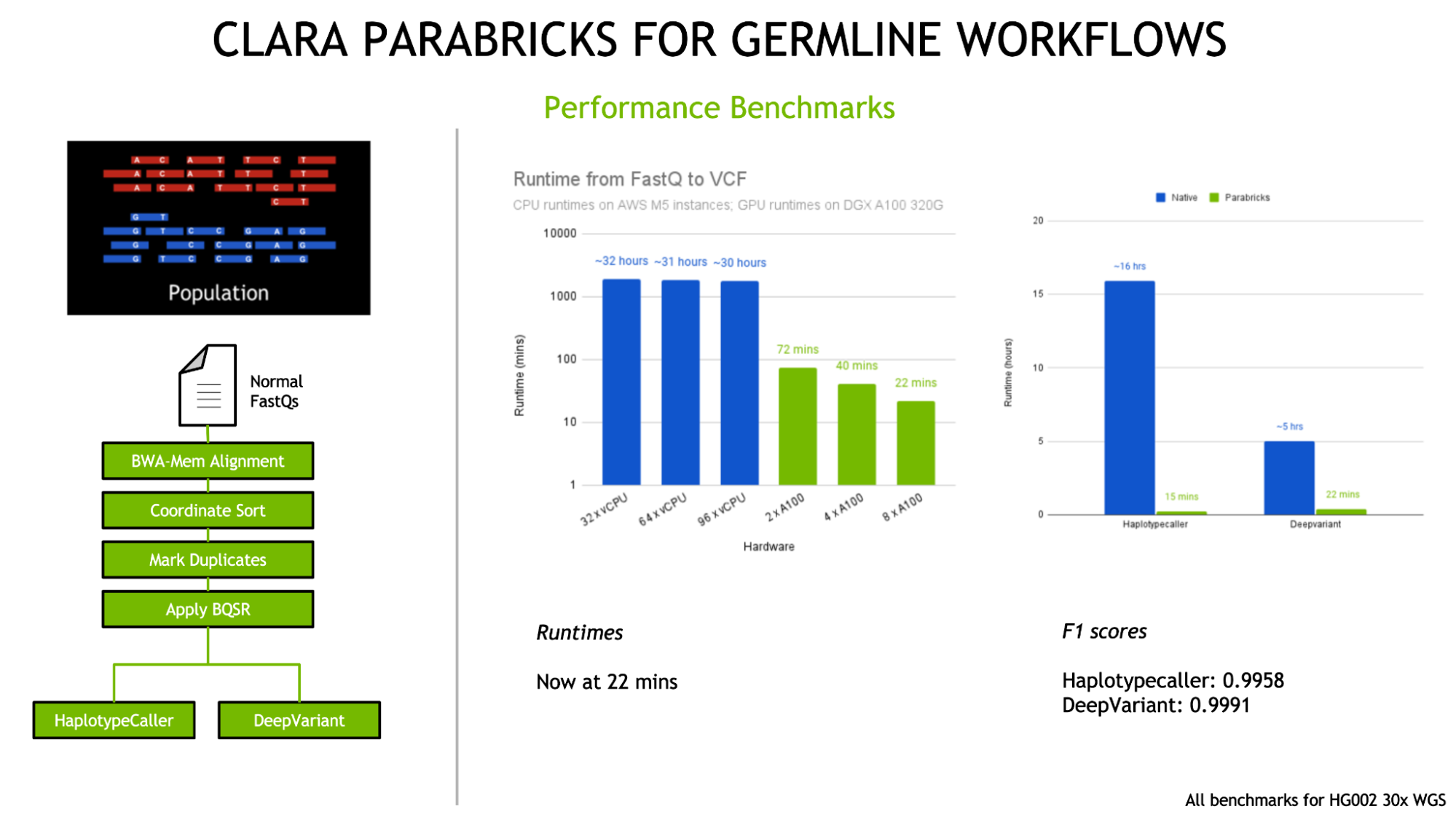
To date, Clara Parabricks has demonstrated 60x accelerations for state-of-the-art bioinformatics tools for whole genome workflows (end-to-end analysis in 22 minutes) and exome workflows (end-to-end analysis in 4 minutes), compared to CPU-based environments. Large-scale sequencing projects and other whole genome studies are able to analyze over 60 genomes/day on a single DGX server while both reducing the associated costs and generating more useful insights than ever before.
Accelerate and simplify gene panel workflows with the support of Unique Molecular Identifiers (UMIs) also known as molecular barcodes.
RNA-Seq support for transcriptome workflows with second gene fusion caller Arriba and RNA-Seq quantification tool Kallisto.
Short tandem repeat (STR) detection with ExpansionHunter.
Integration of the latest versions of germline callers DeepVariant v1.1 and GATK v4.2 with HaplotypeCaller.
A 10x accelerated BAM2FASTQ tool for converting archived data stored as either BAM or CRAM files back to FASTQ. Datasets can be updated by aligning to new and improved references.
Figure 1: Clara Parabricks 3.7 includes a UMI workflow for gene panels, ExpansionHunter for short tandem repeats, inclusion of the DeepVariant 1.1 and GATK 4.2 releases, and Kallisto and Arriba for RNA-Seq workflows.
Clara Parabricks 3.7 accelerates and simplifies gene panel analysis
While whole genome sequencing (WGS) is growing due to large-scale population initiatives, gene panels still dominate clinical genomic analysis. With time being one of the most important factors in clinical care, accelerating and simplifying gene-panel workflows is incredibly important for clinical sequencing centers. By further reducing the analysis bottleneck associated with gene panels, these sequencing centers can return results to clinicians faster, improving the quality of life for their patients.
Cancer samples used for gene panels are commonly derived from either a solid tumor or consist of cell-free DNA from the blood (liquid biopsies) of a patient. Compared to the discovery work in WGS, gene panels are narrowly focused on identifying genetic variants in known genes that either cause disease or can be targeted with specific therapies.
Gene panels for inherited diseases are often sequenced to 100x coverage while gene panels in cancer sequencing are sequenced to a much higher depth, up to several 1,000x for liquid biopsy samples. The higher coverage is required to detect lower frequency somatic mutations associated with cancer.
To improve the limit of detection for these gene panels, molecular barcodes or UMIs are used, as they significantly reduce the background noise. This limit of detection is pushed for liquid biopsies, and can include tens of thousands coverage in combination with UMIs to identify those needle-in-the-haystack somatic mutations circulating in the bloodstream. High-depth gene-panel sequencing can reintroduce a computational bottleneck in the required processing of many more sequencing reads.
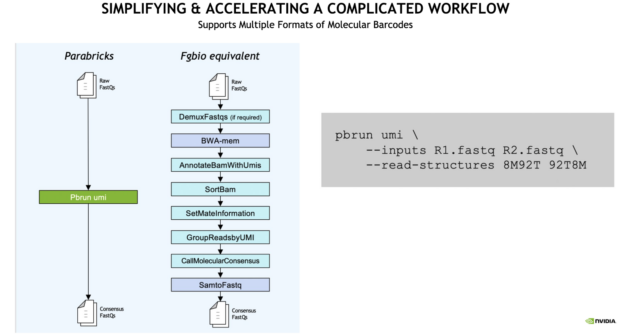
With Clara Parabricks, UMI gene panels can now be processed 10x faster than traditional workflows, generating results in less than an hour.
The analysis workflow is also simplified. From raw FASTQ to consensus FASTQ, a single command line runs multiple inputs compared to the traditional Fulcrum Genomics (Fgbio) equivalent as seen in the example below.
Figure 2: Clara Parabricks 3.7 has simplified and accelerated gene panel UMI workflows compared to the Fgbio equivalent.
RNA-Seq support with Arriba and Kallisto
Just as gene panels are important for sequencing cancer analysis, so too are RNA-Seq workflows for transcriptome analysis. In addition to STAR-fusion, Clara Parabricks v3.7 now includes Arriba, a fusion detection algorithm based on the STARRNA-Seq aligner. Gene fusions, in which two distinct genes join due to a large chromosomal alteration, are associated with many different types of cancer from leukemia to solid tumors.
Arriba can also detect viral integration sites, internal tandem duplications, whole exon duplications, circular RNAs, enhancer hijacking events involving immunoglobulin/T-cell receptor loci, and breakpoints in introns or intergenic regions.
Clara Parabricks v3.7 also incorporates Kallisto, a fast RNA-Seq quantification tool based on pseudo-alignment that identifies transcript abundances (aka gene expression levels based on sequencing read counts) from either bulk or single-cell RNA-Seq datasets. Alignment of RNA-Seq data is the first step of the RNA-Seq analysis workflow. With tools for transcript quantification, read alignment, and fusion calling, Clara Parabricks 3.7 now provides a full suite of tools to support multiple RNA-Seq workflows.
Short tandem repeat detection with ExpansionHunter
To support genotyping of short tandem repeats (STRs) from short-read sequencing data, ExpansionHunter support has been added to Parabricks v3.7. STRs, also referred to as microsatellites, are ubiquitous in the human genome. These regions of noncoding DNA have accordion-like stretches of DNA containing core repeat units between two and seven nucleotides in length, repeated in tandem, up to several dozen times.
STRs are extremely useful in applications such as the construction of genetic maps, gene location, genetic linkage analysis, identification of individuals, paternity testing, population genetics, and disease diagnosis.
There are a number of regions in the human genome consisting of such repeats, which can expand in their number of repetitions, causing disease. Fragile X Syndrome, ALS, and Huntington’s Disease are well-known examples of repeat-associated diseases.
ExpansionHunter aims to estimate sizes of repeats by performing a targeted search through a BAM/CRAM file for sequencing reads that span, flank, and are fully contained in each STR.
The addition of somatic callers and support of archived data
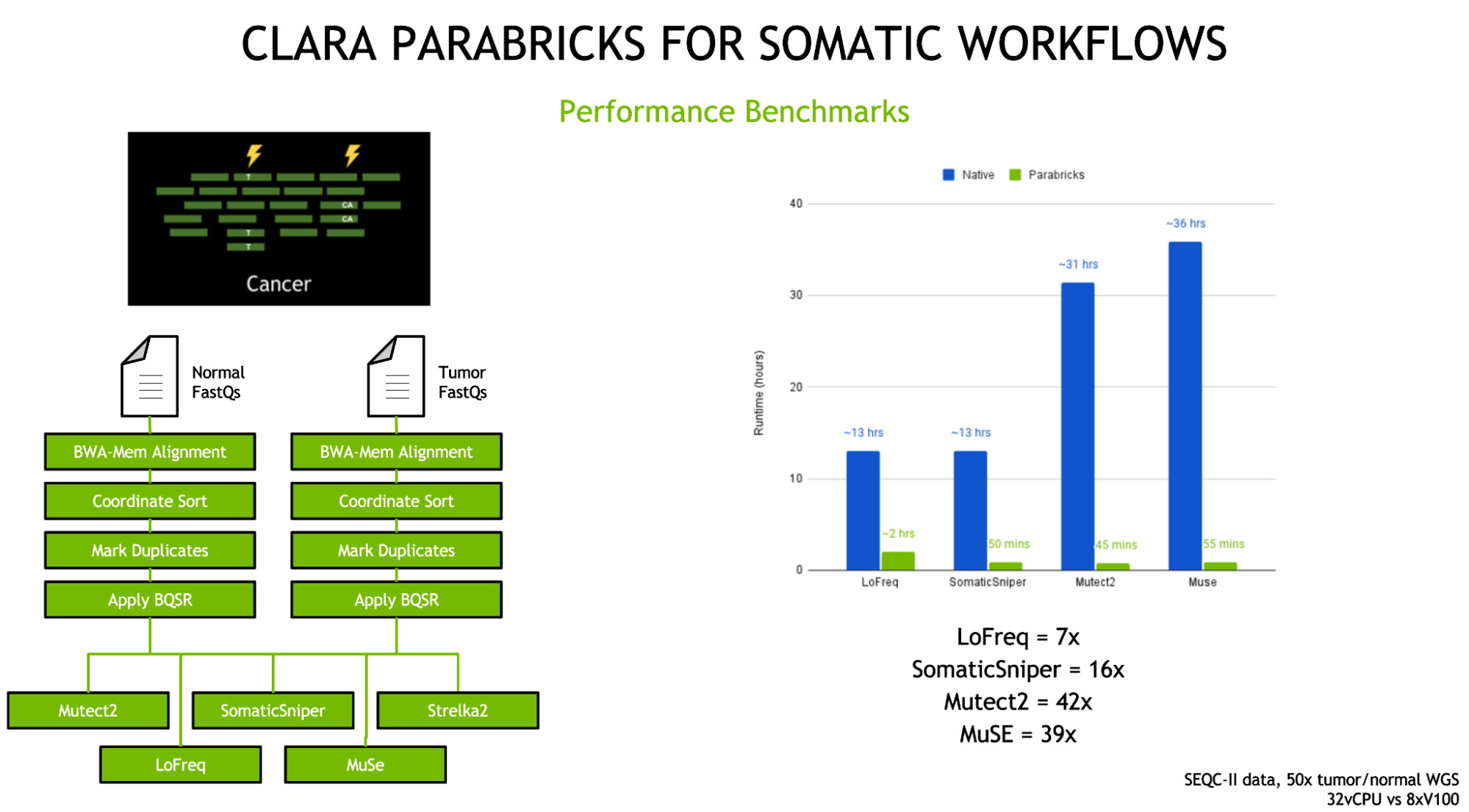
The previous releases of Clara Parabricks in 2021 brought a host of new tools, most importantly the addition of five somatics callers—MuSE, LoFreq, Strelka2, Mutect2, and SomaticSniper—for comprehensive cancer genomic analysis.
Figure 3: Clara Parabricks v3.6.1 and 3.7 includes five somatic callers for comprehensive accelerated cancer genomic analysis—Muse, LoFreq, Strelka2, Mutect2, and SomaticSniper.
In addition, tools were added to take advantage of archived data in scenarios when original FASTQ files were deleted to save storage space. BAM2FASTQ is an accelerated version of GATK Sam2fastq, which converts an existing BAM or CRAM file to a FASTQ file. This allows users to realign sequencing reads to a new reference genome which will enable more variants to be called, along with providing researchers capabilities to normalize all their data to the same reference. Using the existing fq2bam tool, Clara Parabricks can realign reads from one reference to another in under 2 hours for a 30X whole genome.
Figure 4: Clara Parabricks v3.7 has added the latest releases of germline variant callers DeepVariant 1.1 and GATK 4.2 with HaplotypeCaller.
Try out GPU-accelerated NVIDIA Clara Parabricks genomic analysis tools for your germline, cancer, and RNA-Seq analysis workflows with a free with a 90-day evaluation trial license.
Online commerce has rocketed to trillions of dollars worldwide in the past decade, serving billions of consumers. Behind the scenes of this explosive growth in online sales is personalization driven by recommender engines. Recommenders make shopping deeply personalized. While searching for products on e-commerce sites, they find you. Or suggestions can just appear. This wildly Read article >
Hello, I’m currently working through the tensorflow agent and bandit library/tutorials. But I can’t answer completely myself what the difference between the driver and the trainer is. Is it basically the trainer is training the agent and for example his neuronal network for the (approximately) perfect policy in a given environment? And the the driver is just the execution of a given policy without any regards to optimizing it along the way ?
Does anyone know if it’s possible to compile tensorflow lite (C api) as a static library, and more specifically for iOS?
I have a C library that needs to interact with tensorflow lite, which I want to integrate into an iOS application, but iOS only accepts static libraries
Didn’t see that the imgDimentions on the trainer was set to 225,225,1 but the detector was trying to find 255,255,1 images
its been a long day!
Hi all, me again haha
Ive run into an error that has stumped me for about an hour;
Im trying to train a TensorFlow modal to detect a British one pence coin and a British 2 pence coin, while the model trained, when it detects the objects it crashes and gives me this error:
ValueError: Input 0 of layer “sequential” is incompatible with the layer: expected shape=(None, 225, 225, 1), found shape=(None, 255, 255, 1)
its finding the shape its supposed to find, but it says its not? Any advice would be greatly appreciated!
I recently started doing some smaller TF projects and decided that it’s a good time to get some more formal training in the area and with TF.
I found a few interesting resources and was wondering if any of you have completed them and what your thought are? Are there any great resources I am missing on this list?
For background, I am most interested in time series analysis, medical AI, and TF on embedded devices like the Google Coral but think a combination of a broad course + one specialized on these areas would be best.
I’m starting to prepare for the certification and from what I know jupyter notebooks are only editable in pycharm professional edition. The exercises in the course im taking (DeepLearning.AI TensorFlow Developer Professional Certificate) are in ipynb format.
So I’m a little confused if the test will be done in .ipynb or .py format? If it is in .ipynb, will I need to get pycharm professional? Thanks in advance!
New CUDA 11.6 Toolkit is focused on enhancing the programming model and performance of your CUDA applications.
NVIDIA announces the newest release of the CUDA development environment, CUDA 11.6. This release is focused on enhancing the programming model and performance of your CUDA applications. CUDA continues to push the boundaries of GPU acceleration and lay the foundation for new applications in HPC, visualization, AI, ML and DL, and data science.
CUDA 11.6 has several important features. This post offers an overview of the key capabilities:
GSP driver architecture now default on Turing and Ampere GPUs
New API to allow disabling nodes in instantiated graph
The GSP driver architecture is now the default driver mode for all listed Turing and Ampere GPUs. The older driver architecture is supported as a fallback. For more information, see R510 Driver Readme.
Instantiated Graph Node API additions
We added a new API, cudaGraphNodeSetEnabled, to allow disabling nodes in an instantiated graph. Support is limited to kernel nodes in this release. A corresponding API, cudaGraphNodeGetEnabled, allows querying the enabled state of a node. We’ve also added the ability to disable NULL kernel graph node launches.
128-bit integer support
CUDA 11.6 includes the full release of 128-bit integer (__int128) data type, including compiler and developer tools support. The host-side compiler must support the __int128 type to use this feature.
Cooperative groups namespace
The cooperative groups namespace has been updated with new functions to improve consistency in naming, function scope, and unit dimension and size.
Implicit Group/Member
Threads
Blocks
thread_block::
dim_threads num_threads thread_rank thread_index
(Not needed)
grid_group::
num_threads thread_rank
dim_blocks num_blocks block_rank block_index
Table 1. New functions in cooperative groups namespace
CUDA compiler
Added -arch=native compilation option to target installed GPUs during compilation. This extends the existing -gencode=arch=compute_xx,code=sm_xx architecture specification
Add the ability to create PTX files from nvlink
Deprecated features
The cudaDeviceSynchronize() used for on-device fork and join parallelism is deprecated in preparation for a replacement programming model with higher performance. These functions continue to work in this release, but the tools emit a warning about the upcoming change.
CentOS Linux 8 has reached End-of-Life on Dec 31, 2021, and support for this OS is now deprecated in the CUDA Toolkit. CentOS Linux 8 support will be completely removed in a future release.
I’m trying to make a GAN model to reconstruct one image from another. So I have two MRI images I put one example of these images, which my input is the top one and the target is the bottom one. My problem here is that I’m trying to create a custom loss function that would penalize pixels that are from the background that are not inside of the brain. A professor told me about masking the image inside of the loss function so I’m kinda lost :/. Can anyone help me ?

 GANcraft is a hybrid neural rendering pipeline to represent large and complex scenes using Minecraft.
GANcraft is a hybrid neural rendering pipeline to represent large and complex scenes using Minecraft.




 The GPU-accelerated Clara Parabricks v3.7 release brings support for gene panels, RNA-Seq, short tandem repeats, and updates to GATK 4.2 and DeepVariant 1.1.
The GPU-accelerated Clara Parabricks v3.7 release brings support for gene panels, RNA-Seq, short tandem repeats, and updates to GATK 4.2 and DeepVariant 1.1.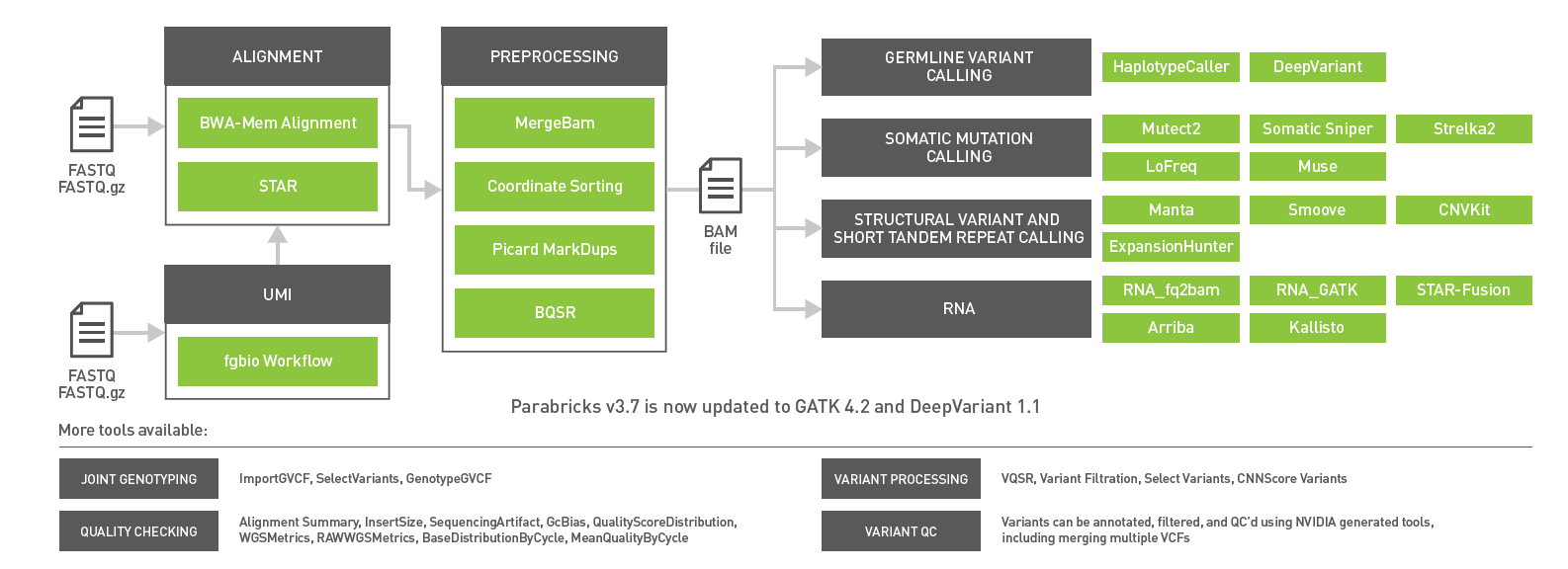

") New CUDA 11.6 Toolkit is focused on enhancing the programming model and performance of your CUDA applications.
New CUDA 11.6 Toolkit is focused on enhancing the programming model and performance of your CUDA applications.{kind=link}